

New Primary Haemophagocytic Lymphohistiocytosis treatments 2024
New Primary Haemophagocytic Lymphohistiocytosis Treatments 2024
Primary Hemophagocytic Lymphohistiocytosis (HLH) is a rare but potentially life-threatening immune disorder that typically affects infants and young children, although it can also present in adults. It is characterized by an excessive and inappropriate immune response, leading to severe inflammation and tissue damage. The symptoms of HLH can be nonspecific and include persistent fever, enlargement of the liver and spleen, cytopenias, and neurological abnormalities. Diagnosis is based on clinical criteria and laboratory tests, including elevated levels of triglycerides, ferritin, and soluble interleukin-2 receptor, as well as evidence of hemophagocytosis in bone marrow, spleen, or lymph nodes. Genetic testing can confirm the diagnosis, as familial forms of HLH are linked to mutations in genes responsible for the function of natural killer cells and cytotoxic T lymphocytes.
Treatment options for Primary HLH are aimed at controlling the hyperinflammatory response and preparing the patient for potential hematopoietic stem cell transplantation (HSCT), which is currently the only curative treatment. Initial therapy often includes immunosuppressive drugs such as steroids (e.g., dexamethasone) and etoposide, an antineoplastic agent. In some cases, cyclosporine A, an immunomodulating agent, is also used to control the disease. Biological therapies, like the monoclonal antibody against CD52 (alemtuzumab), may be considered in refractory cases or as a bridge to transplantation. Due to the complexity of the disease and the potential for serious side effects, treatment should be managed by a multidisciplinary team with expertise in HLH and HSCT.

Treatment options
| Treatment option | Estimated cost | Efficacy | Eligibility |
|---|---|---|---|
| Chemotherapy (e.g., Etoposide) | Varies | Can be effective as part of HLH-94 protocol | Typically used in combination with other treatments; eligibility depends on patient's overall health and specific HLH criteria |
| Immunotherapy (e.g., Antithymocyte globulin) | Varies | Used to suppress the immune system; efficacy varies | Considered for patients who do not respond to initial chemotherapy |
| Corticosteroids (e.g., Dexamethasone) | Relatively low | Often effective in reducing inflammation | Widely eligible; often part of initial treatment protocol |
| Stem cell transplant | High | Potentially curative | Eligibility depends on the availability of a suitable donor and patient's ability to tolerate the procedure |
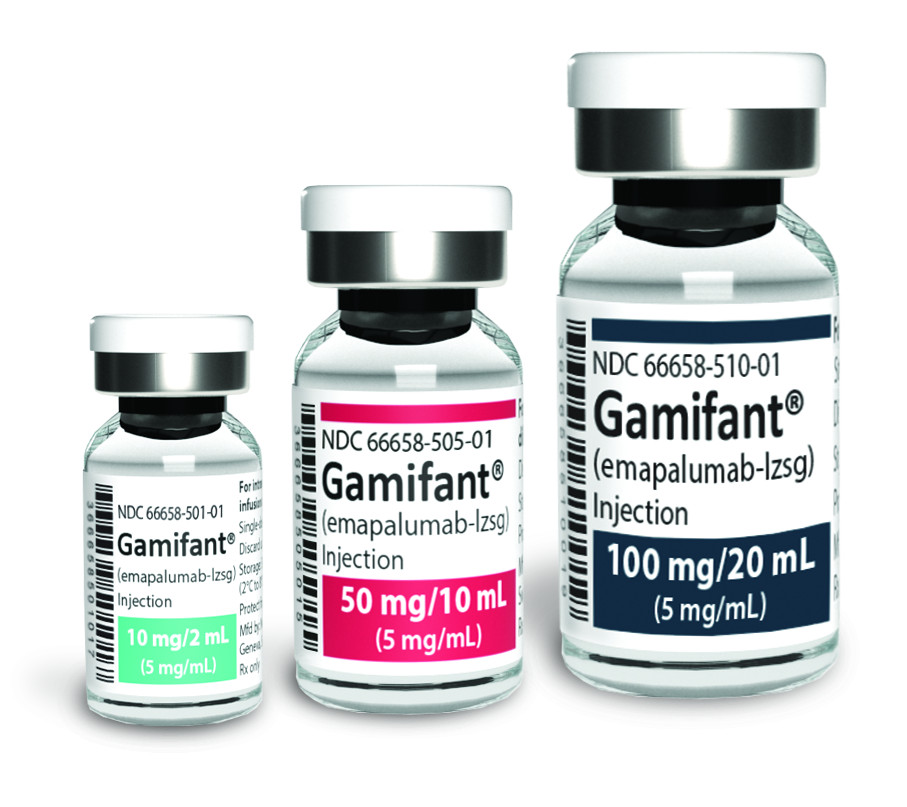
| Gamifant (Emapalumab) | Very high | Approved by the FDA for patients with refractory, recurrent, or progressive disease or intolerance to conventional HLH therapy | FDA-approved for primary HLH with specific genetic mutations |
| Experimental treatments (e.g., Ruxolitinib) | Varies; often covered by research funds | Varies; under investigation | Typically available only through clinical trials; eligibility depends on trial inclusion criteria |
Treatments options in detail
First-Line Treatment for Primary Hemophagocytic Lymphohistiocytosis (HLH)
Primary Hemophagocytic Lymphohistiocytosis (HLH) is a life-threatening immunologic condition that requires prompt treatment. The first-line treatment for HLH is typically a combination of immunosuppressive and chemotherapeutic agents. The HLH-94 protocol, which includes etoposide (VP-16), dexamethasone, and, in some cases, cyclosporine A, is the most commonly used regimen. Etoposide is a chemotherapeutic agent that helps to control the excessive immune activation by targeting rapidly dividing cells, including activated immune cells. Dexamethasone is a corticosteroid that reduces inflammation and immune system activity. Cyclosporine A is an immunosuppressive drug that further helps to control the hyperinflammatory response.
Intrathecal Therapy
For patients with central nervous system (CNS) involvement, intrathecal therapy with methotrexate and corticosteroids may be administered. This involves the direct injection of medication into the cerebrospinal fluid to reduce inflammation and immune cell activation in the CNS, which is a common and serious complication of HLH.
Hematopoietic Stem Cell Transplantation (HSCT)
After initial stabilization with medical therapy, hematopoietic stem cell transplantation (HSCT) is considered the only curative treatment for primary HLH. HSCT aims to replace the defective immune system with a healthy one from a compatible donor. The success of HSCT largely depends on the disease's state at the time of transplant and the patient's overall health.
Immunoglobulin Therapy
Intravenous immunoglobulin (IVIG) is sometimes used as adjunctive therapy in HLH. IVIG is thought to work by providing normal antibodies that can help to regulate the immune system, although its exact mechanism in HLH is not fully understood.
Biologic Therapy with Gamifant (Emapalumab-lzsg)
Gamifant (emapalumab-lzsg) is a monoclonal antibody that targets interferon gamma (IFNγ), a key cytokine involved in the inflammatory process of HLH. It is the first and only FDA-approved treatment specifically for primary HLH in patients with refractory, recurrent, or progressive disease or intolerance to conventional HLH therapy. Gamifant works by neutralizing IFNγ, thereby reducing the hyperinflammation associated with HLH. It is typically used when conventional treatments have failed or are not tolerated.
Antithymocyte Globulin (ATG)
Antithymocyte globulin (ATG) is another immunosuppressive agent that may be used in the treatment of HLH. It is derived from the serum of animals immunized with human thymocytes and works by depleting T-cells, which are often hyperactive in HLH. ATG is generally considered when the response to standard therapy is inadequate.
Supportive Care
Supportive care is a critical component of HLH treatment and includes management of symptoms and complications. This may involve transfusions of red blood cells and platelets, antibiotics or antifungals for infections, and management of metabolic and electrolyte disturbances. Fever and pain are managed with appropriate medications, and organ functions are closely monitored and supported as needed.
Experimental Treatments
Research into new treatments for HLH is ongoing. Experimental approaches may include the use of novel biologic agents that target specific pathways involved in the hyperinflammatory process. Clinical trials are evaluating the safety and efficacy of these new treatments. Patients interested in experimental therapies should discuss the potential risks and benefits with their healthcare provider and may consider enrolling in a clinical trial.
Treatments Not Approved by the FDA
There are treatments used for HLH that are not specifically approved by the FDA for this condition but may be used off-label or are available through clinical trials. These include various chemotherapeutic agents, immunosuppressants, and biologic therapies that have shown promise in controlling the disease. For example, rituximab, an anti-CD20 monoclonal antibody, has been used in cases where HLH is associated with underlying lymphoproliferative disorders. Off-label use of medications should be guided by a specialist with experience in treating HLH, and the potential benefits must be weighed against the risks.
Conclusion
The treatment of primary HLH is complex and requires a multidisciplinary approach. Early intervention with the HLH-94 protocol is critical, followed by HSCT for a curative intent. Gamifant provides a targeted treatment option for those with refractory disease. Supportive care remains a cornerstone of management, and experimental and off-label treatments offer additional avenues for those who do not respond to standard therapies. Ongoing research and clinical trials continue to expand the understanding and treatment options for this challenging condition.
Symptoms
Symptoms of Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis (HLH) is a rare but severe immune disorder. It is characterized by a hyperinflammatory response due to the overactivation of lymphocytes and histiocytes, which can lead to tissue damage and organ failure. The most common symptoms of primary HLH are prolonged fever and splenomegaly. Fever in HLH is typically high-grade and persistent, often not responding to standard antipyretic treatments. Splenomegaly, which is the enlargement of the spleen, can be detected through physical examination or imaging studies.
Cytopenias, or the reduction in the number of blood cells, is another hallmark of primary HLH. Patients may present with anemia, which can cause fatigue, pallor, and shortness of breath. Thrombocytopenia, or low platelet counts, can lead to easy bruising, bleeding gums, and petechiae (small red or purple spots on the body caused by minor bleeding). Neutropenia, a decrease in the number of neutrophils, can increase the risk of infections.
Hepatomegaly, or the enlargement of the liver, is also frequently observed in primary HLH patients. This may be accompanied by elevated liver enzymes, indicating liver dysfunction. Jaundice, or yellowing of the skin and eyes, may occur as a result of liver impairment.
Lymphadenopathy, the swelling of lymph nodes, is common in individuals with primary HLH. This can be felt as lumps under the skin, particularly in the neck, armpits, and groin areas. Patients may also experience general symptoms such as weight loss, night sweats, and general malaise.
Neurological symptoms are present in a significant number of primary HLH cases. These can include irritability, seizures, ataxia (lack of muscle control), headaches, and alterations in consciousness, ranging from confusion to coma. Meningitis, which is the inflammation of the membranes covering the brain and spinal cord, can also develop.
Rash or skin manifestations, such as a generalized maculopapular rash, can be seen in primary HLH. These skin changes may be nonspecific but can be indicative of the underlying systemic inflammation.
Hypertriglyceridemia and hypofibrinogenemia are often detected through laboratory tests. Elevated triglyceride levels and reduced fibrinogen levels are indicative of the metabolic disturbances that occur in HLH.
Hyperferritinemia, or high levels of ferritin in the blood, is a common laboratory finding in primary HLH and reflects the intense inflammatory process. Ferritin levels are often markedly elevated and can be used as a diagnostic marker for the disease.
Hemophagocytosis, which is the phagocytosis of blood cells by histiocytes, is a characteristic feature of HLH, although it is not always present. This can be observed in bone marrow, spleen, or lymph node biopsies.
Coagulation abnormalities are also common in primary HLH, with patients experiencing prolonged clotting times, which can lead to an increased risk of bleeding. Disseminated intravascular coagulation (DIC) may occur in severe cases.
Patients with primary HLH may also exhibit signs of immune dysregulation, such as hypogammaglobulinemia or hypergammaglobulinemia, reflecting abnormal immunoglobulin levels. This can contribute to the increased susceptibility to infections seen in HLH.
Respiratory symptoms, including coughing and difficulty breathing, can develop as a result of lung involvement. Infiltrates may be seen on chest radiographs, indicating inflammation or infection.
Gastrointestinal symptoms such as diarrhea, vomiting, and abdominal pain can also occur, further complicating the clinical picture. These symptoms may result from gastrointestinal tract involvement or the effects of systemic inflammation.
It is important to note that the symptoms of primary HLH can mimic those of other conditions, making diagnosis challenging. A high index of suspicion is necessary, particularly in patients with persistent fever, cytopenias, and organomegaly, to prompt further investigation and confirm the diagnosis of primary HLH.
Due to the severity and rapid progression of the disease, early recognition and treatment of primary HLH are critical. The constellation of symptoms, along with laboratory and histopathological findings, guide the diagnostic process and subsequent management of the condition.
Cure
Current Treatment Approaches for Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis (HLH) is a severe systemic inflammatory syndrome that can be fatal if left untreated. As of the current medical understanding, there is no outright "cure" for primary HLH, but there are treatment protocols that aim to manage the condition, reduce symptoms, and improve survival rates. The mainstay of treatment for primary HLH involves a combination of immunosuppressive therapy, chemotherapy, and biological therapy to control the hyperinflammation.
Immunosuppressive and Chemotherapy Regimens
The initial treatment for primary HLH typically includes the use of steroids, such as dexamethasone, to suppress the immune system's overactivity. Alongside steroids, etoposide (VP-16) is commonly used for its chemotherapeutic effects to eliminate activated immune cells. This combination aims to control the disease and prepare the patient for potential hematopoietic stem cell transplantation (HSCT).
Hematopoietic Stem Cell Transplantation (HSCT)
HSCT is currently the closest approach to a potential cure for primary HLH and is generally recommended for patients with genetic forms of the disease. The procedure involves the transplantation of stem cells from a healthy donor to replace the patient's defective immune system. HSCT has been shown to improve long-term survival rates significantly, but it carries risks such as graft-versus-host disease, infection, and transplant rejection.
Biological Therapy
Biological therapies, such as the use of the interleukin-1 receptor antagonist anakinra or the anti-thymocyte globulin (ATG), have been used in the treatment of primary HLH to target specific pathways in the immune response. These therapies can be particularly useful in patients who are not candidates for HSCT or in those who need to control the disease before transplantation.
Supportive Care
Supportive care is critical in the management of primary HLH and includes measures to control infections, manage organ dysfunctions, and maintain the patient's general health. Intravenous immunoglobulin (IVIG) may also be used to provide passive immunity to patients with severe infections.
Off-Label and Investigational Therapies
Off-label use of medications not specifically approved for primary HLH but that have shown promise in managing the disease includes drugs like ruxolitinib, a JAK1/2 inhibitor, which has been used in cases refractory to conventional treatment. Clinical trials are ongoing to evaluate the efficacy and safety of these and other novel agents in the treatment of primary HLH.
Genetic Therapies
Research into genetic therapies, including gene editing and gene therapy, is ongoing and may offer hope for a cure in the future. These experimental treatments aim to correct the underlying genetic defects that cause primary HLH. However, these therapies are still in the early stages of development and are not yet available outside of clinical trials.
Conclusion on Cure
While there is currently no definitive cure for primary Hemophagocytic Lymphohistiocytosis, advances in treatment, particularly HSCT, offer the best chance for long-term remission and survival. The management of primary HLH is complex and requires a multidisciplinary approach, often involving specialists in immunology, hematology, and transplant medicine. Continuous research and clinical trials are essential to improve outcomes and possibly discover a cure in the future.
Access Primary Haemophagocytic Lymphohistiocytosis medicines today
If Primary Haemophagocytic Lymphohistiocytosis medicines are not approved or available in your country (e.g. due to supply issues), you can access them via Everyone.org.
How Everyone.org works

Make an enquiry
Choose the medicine you want to access, answer a couple of questions, and upload your prescription to speed things up. We’ll get back to you within 24 hours.


Make an enquiry
Choose the medicine you want to access, answer a couple of questions, and upload your prescription to speed things up. We’ll get back to you within 24 hours.

Breeze through the paperwork
We'll guide you through the required documents for importing unapproved medicine, ensuring you have all the necessary information.


Get a personalized quote
We’ll prepare a quote for you, including medicine costs and any shipping, administrative, or import fees that may apply.


Receive your medicine
Accept the quote and we’ll handle the rest - sourcing and safely delivering your medicine.

Some text on this page has been automatically generated. Speak to your physician before you start a new treatment or medication.
Let's talk
If you have any questions, call us or send us a message through WhatsApp or email:
Contact us